
17/12/2021
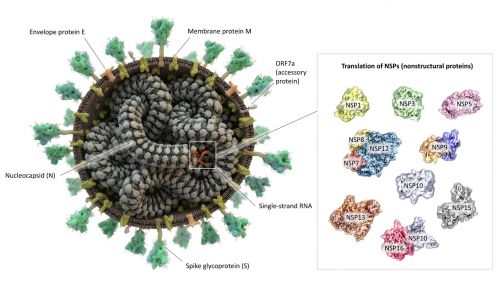
The Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) is the causative agent of the Coronavirus disease 2019 (COVID-19), which to-date accounts for almost 5 million deaths globally. The diffusion of the COVID-19 pandemic has produced emerging variants, which are tracked through sequencing efforts surpassing that of any other pathogen. Understanding the molecular basis of SARS-CoV-2 infection is a key priority since the emergence of the COVID-19 disease, because a detailed, mechanistic understanding of the infectivity and replication mechanisms of the virus is necessary to tackle it through targeted drugs and vaccines resistant to escape variants.
“SCoV2-MD enables the interactive analysis of the deposited trajectories through a web interface, which enables users to search by viral protein, isolate, phylogenetic attributes, or specific point mutation” said Dr. Jana Selent, of the Hospital del Mar Medical Research Institute, one of the leading authors of the paper.
Large-scale computational experiments, known as molecular dynamics (MD) simulations, enable the study of transient phenomena occurring at molecular scales, at temporal and spatial resolutions unavailable to most experimental techniques.
“With SCoV2-MD we link the time-resolved viral proteome dynamics with information on existing SARS-CoV-2 variants, their phylogeny, and the corresponding individual isolates. This allows for the first time to extensively evaluate the structural impact of variants taking into account protein dynamics” added Dr. Toni Giorgino, of the Institute of Biophysics of the National Research Council, co-lead author of the paper presenting the work, which was published as a "Breakthrough Article" in the Nucleic Acids Research journal.
MD data are highly relevant to understand the functional dynamics of the viral proteome which cannot often be deduced from the static structure that has been experimentally solved. In addition, they help rationalize the structural/functional impact of sequence variability in the viral proteome. This is particularly useful when the relationship between mutation location and activity is not obvious (e.g. the mutation is distant from the protein’s active center).
In SCov2-MD each mutation can be analyzed interactively combining static (e.g. a variety of amino acid substitution penalties) and dynamic (time-dependent data derived from the dynamics of the local geometry) scores. Dynamic scores can be computed on the basis of nine non-covalent interaction types, including steric properties, solvent accessibility, hydrogen bonding, and other types of chemical interactions. Where available, experimental data such as antibody escape and change in binding affinities from deep mutational scanning experiments are also made available. All metrics can be combined to build predefined or custom scores to interrogate the impact of evolving variants on protein structure and function.
SCoV2-MD is an opportunity to demonstrate the commitment to an inclusive, collaborative and interdisciplinary science that has significant implications in the understanding of pathogenic mechanisms, as well as spin-offs in the discovery of urgent treatments.
Paper details: Torrens-Fontanals, M., Peralta-García, A., Talarico, C., Guixà-González, R., Giorgino, T., Selent, J. SCoV2-MD: a database for the dynamics of the SARS-CoV-2 proteome and variant impact predictions. Nucleic Acids Research (2021). doi:10.1093/nar/gkab977
Per informazioni:
Toni Giorgino
CNR - Istituto di biofisica
via Celoria, 26 - 20133 Milano
toni.giorgino@cnr.it
Vedi anche: